|
Case
Report
Diagnostic
Dilemma in a Young Female with
Osteolytic Lesion of Long Bone
Authors:
B Yuva Jyothi,
Junior Resident (Pathology), Department
of Laboratory Medicine, Command Hospital
Airforce, Bangalore, Karnataka, India,
Seerat Pal, Associate
Professor, Dept of Laboratory Medicine,
Command Hospital Airforce, Bangalore,
Karnataka, India,
Ritu Mehta, Professor,
Dept of Laboratory Medicine, Command
Hospital Airforce, Bangalore, Karnataka,
India,
Surjeet Dwivedi,
Professor, Dept of Surgery, Armed Forced
Medical College, Pune, India,
Arun Ravi John,
Associate Professor, Dept of Nuclear
Medicine, Command Hospital Airforce,
Bangalore, Karnataka, India,
Abhishek Kumar Sharma,
Assistant Professor, Department of
Medicine, Command Hospital Airforce,
Bangalore, Karnataka, India,
Deepti Mutreja,
Professor and Head (Pathology),
Department of Laboratory Medicine,
Command Hospital Airforce, Bangalore,
Karnataka, India.
Address for
Correspondence
Dr Seerat Pal,
Associate Professor,
Dept of Laboratory Medicine,
Command Hospital Airforce,
Bangalore, Karnataka - 560007, India.
E-mail:
seeratpalsharma@gmail.com.
Citation
Jyothi BY, Pal S, Mehta
R, Dwivedi S, John AR, Sharma AK,
Mutreja D. Diagnostic Dilemma in a Young
Female with Osteolytic Lesion of Long
Bone. Online J Health Allied Scs.
2026;25(1):3. Available at URL:
https://www.ojhas.org/issue97/2026-1-3.html
Submitted:
Jan
10, 2026; Accepted: Apr 5, 2026;
Published: Apr 25, 2026
|
|
|
|
|
Introduction
Immunoglobulin
G4-related disease (IgG4-RD) is an immune-mediated
fibroinflammatory disease which can affect
multiple organ systems. This disease is usually
associated with high serum IgG4 levels and
specific histopathological features.
The concept of
IgG4-RD was first introduced by Hamano et al. in
2001. It is associated with increased serum IgG4
levels in patients with autoimmune pancreatitis
(AIP)[1]. The etiology and triggering factors are
still unknown. The condition occurs most often in
middle-aged or older men. A single study from
Japan reported a low male to female ratio of 1:
0.77 [2]. Involvement of almost all anatomical
regions has been reported, but most commonly
affected areas are the pancreas, lacrimal gland,
salivary gland, retroperitoneum, orbit, lymph
nodes, kidneys, and lungs [1]. IgG4-RD is often
discovered incidentally during radiological or
histopathological examination of tissues. The
diagnosis is made by combined evaluation of
clinical, radiological, and histopathological
findings. The course of the disease is usually
characterized by remission and recurrent attacks,
which can lead to fibrosis, destructive tissue
damage, and if not promptly treated, it can cause
organ failure. It also causes compression
findings, secondary sclerosis, and obstruction due
to neoplastic lesions [3,4]. Correct
identification is essential to avoid unnecessary
major surgery and to institute corticosteroid
therapy [2].
Case History
A 31 years old
female with no co-morbidities presented with
complaints of pain in the left thigh for 1 month,
difficulty in weight bearing and pain while
walking for 5 days duration, which was insidious
in onset, gradually progressive, aggravated on
weight bearing associated with radiating pain to
back and left thigh. Local examination revealed
tenderness and swelling in the middle 1/3rd of
left thigh. There was no local rise of temperature
and overlying skin was normal. Terminal flexion
movement was painful at left hip and left knee
joint. She was unable to perform straight leg
raising test but no neurovascular deficit was
noted.
X ray left thigh
showed a lytic lesion in upper 1/3rd of left femur
with unicortical pathological fracture (Fig 1a).
The CT angiogram and MRI showed an expansile lytic
lesion measuring 9 x 3 cm involving the proximal
shaft of left femur with adjacent marrow and soft
tissue edema. Minimal fluid collection with mild
soft tissue thickening was noted adjacent to the
lesion Fig 1b). PET scan revealed a well-defined
osteolytic lesion measuring 2.5 x 2.6 x 11.1 cm in
the mid and distal 1/3rd of shaft of
femur with significant cortical thinning and
cortical erosion noted with increased uptake (Fig
2 a-f). The clinical differentials considered were
mainly Giant cell tumour, infective osteomyelitis
and malignancy. Laboratory investigations showed
mild anemia (Hb – 9 gm/dl) with neutrophilic
leukocytosis (TLC- 12,400/cumm) with mild left
shift of neutrophils. Initial imaging guided
biopsy from the osteolytic lesion showed only
xanthogranulomatous inflammation with histiocytes
and dense lymphoplasmacytic infiltrate extending
between skeletal muscles with areas of necrosis
and granulation tissue (Fig 3 a-c).
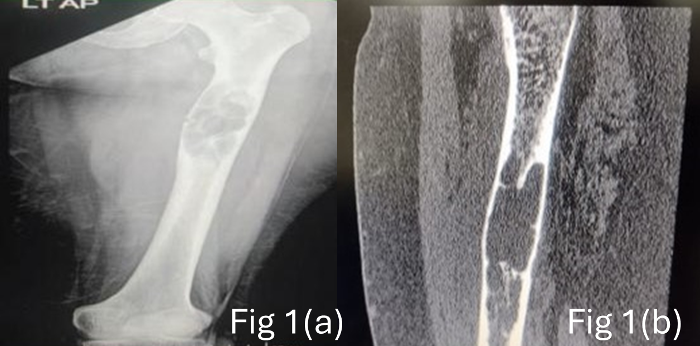
|
| Figure
1(a): X-ray showing lytic lesion in upper
1/3rd of left femur with unicortical
pathological fracture (b) CT Angiogram
-Well defined, lobulated, eccentrically
located lytic lesion in diaphysis of left
femur measuring 23 x 27 x 88 m with
cortical destruction. |

|
| Fig 2 (a) Maximum
Intensity Projection (MIP) image of 18
F-Fluorodeoxy Glucose (FDG) PET showing
focus of increased tracer concentration in
the region of left thigh. Fig 2 (b, c, d):
Fused coronal, (2e) axial image, (2 f)
fused sagittal image of 18 F-FDG PET-CT
showing a metabolically active soft tissue
component in the left thigh adjacent to an
expansile lytic lesion involving the upper
and mid 1/3rd diaphysis of left
femur with significant cortical thinning
and endosteal scalloping. |

|
| Fig 3 (a) dense
inflammatory infiltrate comprising of
neutrophils, lymphocytes, plasma cells,
giant cells and histiocytes; (b) Storiform
type fibrosis; (c) Obliterative phlebitis. |
Patient underwent
currettage with bone grafting and open reduction
internal fixation by nailing. The curettage bony
tissue was sent for histopathology which showed
skeletal muscle bundles infiltrated by numerous
plasma cells, lymphocytes, neutrophils and
histiocytes. The stromal tissue showed degenerated
bony bits with surrounding areas of storiform
fibrosis and occasional blood vessels showing
obliterative phlebitis (Fig 3 a-c). Areas of
necrosis along with xantho-granulomatous changes
& foreign body giant cell reaction was
also noted. No frank evidence of malignancy was
noted.
Based on the
microscopic findings, the probability of infective
osteomyelitis was considered. Tuberculosis was
ruled out as no granulomas or any caesous necrosis
was seen. No Acid-Fast Bacilli was noted on ZN
stain. Fungal cause was ruled out by Pas stain and
Grocott stain showing no fungal elements.
Since the tissue did
not exhibit any typical histomorphology that would
support a giant cell tumor, the clinical suspicion
of a giant cell tumor was ruled out. IHC for CD1a
was negative, ruling out Langan's cell
histiocytosis. Negative ALK expression on IHC
excluded inflammatory myofibroblastic tumor while
negative CD34 expression excluded Dermato
Fibrosarcoma Protuberans. Sarcoma was ruled out
because there were no malignant osteoid or
chondroid regions, no large cells in the backdrop
of mononuclear cells, and IHC was negative for
vimentin, desmin, calponin, S-100, and BCL2. PANCK
negativity ruled out metastatic deposits from
epithelial malignancy.
At this point, we
thought about the possibility of an IgG4-related
illness, but as there were no publications in the
literature about this condition affecting long
bones, it was seen as a lower differential
diagnosis and was only taken into consideration
after all other options had been ruled out.
Immunohistochemistry revealed increased number of
plasma cells highlighted by CD 138, and these
plasma cells showed no kappa or lambda restriction
highlighting their polyclonal nature. These plasma
cells showed strong positivity with IgG4
(>30/hpf) >40% ratio of IgG and IgG4 plasma
cells. Further serum IgG4 studies showed elevated
levels (282 mg/dl) against a normal of 135 mg/dl.
Discussion
IgG4-related disease
(IgG4-RD) is a fibroinflammatory condition that
affects multiple systems. Three diagnostic
criteria are included in the Comprehensive
diagnostic (CD) criteria for IgG4-RD (revised 2020
version)[5]: organ involvement (diffuse/localized
swelling); serum IgG4 concentration >135 mg/dl;
microscopy demonstrating significant plasma cell
infiltration (>10 IgG4+ cells/HPF) and a
>40% ratio of IgG4+/IgG + cells, along with
fibrosis on biopsy. Cases which met all three
criteria were diagnosed with definite IgG4-RD;
those who met the first and last criteria but did
not have elevated serum IgG4 concentration were
diagnosed with probable IgG4-RD; and those who met
the first two criteria but had negative
histopathology results or no histopathologic
examination were diagnosed with possible
IgG4-RD.[5,6] Organ-specific criteria may be used
to re-diagnose patients with a suspected or
probable diagnosis of IgG4-RD.
Although elevated
serum IgG4 levels is a distinctive finding and
frequently seen in IgG4-RD, it’s not the gold
standard for diagnosing the condition. Due to its
low sensitivity of 51% and low specificity of 60%,
serum IgG4 concentration has been questioned in
few studies.[7] As a result, a serum IgG4 cutoff
level of 135 mg/dl was included in the initial CD
criteria for IgG4-RD along with additional
organ-specific criteria because it was thought to
be a distinct and trustworthy sign predictive of
IgG4-RD, and was included in the original CD
criteria for IgG4-RD as well as other
organ-specific criteria.[5 ]
Dense infiltration
of lymphocytes, plasma cells, and fibrosis are
frequent histopathologic features of IgG4-RD. In
addition to lymphoplasmacytic infiltration,
storiform fibrosis and obliterative phlebitis are
considered to be distinctive and characteristic
features for IgG4-RD.[5,6] Additional pathological
criteria - obliterative phlebitis and storiform
fibrosis were included in the 2020 RCD criteria as
these were common histological findings seen in
IgG4-RD. Obliterative phlebitis is characterized
by the obliteration of the vascular lumen with
inflammatory cells and fibrosis, while storiform
fibrosis is defined as cart wheel arrangement of
spindle-shaped cells.
Recent reviews
emphasize that the pathogenesis of IgG4-RD is
rooted in immune dysregulation, particularly in
interactions between T follicular helper cells,
regulatory T cells, and cytotoxic CD4+ T
lymphocytes. These cellular populations drive both
the inflammatory (proliferative) and fibrotic
phases of disease. While the classic view focused
on a predominant role for IgG4 antibodies, new
data highlights the central contribution of
cytotoxic T cells and their secreted fibrogenic
cytokines, such as IL-4, IL-10, and TGF-β, which
mediate organ fibrosis.[7]
The case presented
here involving the femur adds to the limited cases
of long bone involvement, which is likely the
first reported case in literature. Bone lesions
due to IgG4-RD pose significant diagnostic
challenges because they can radiologically and
histologically mimic infections, malignancies, or
other inflammatory disorders.
In the current case,
features like storiform fibrosis, obliterative
phelbitis and a high proportion of IgG4-positive
plasma cells (40% of all IgG-positive plasma
cells) substantiated the diagnosis after ruling
out all possible infectious and neoplastic causes
on thorough IHC panel and by special stains. In
our study, the presence of elevated serum IgG4
supports systemic activity, though levels are not
exclusively diagnostic since increased serum IgG4
can occur in other conditions.[8]
Epidemiological
trends indicate IgG4-RD is likely underdiagnosed,
especially in its fibrotic phenotype, which can
slowly cause irreversible tissue damage. Bone
involvement remains unusual, with a few documented
cases affecting the cranial, temporal, and long
bones. This rarity can delay recognition and
definitive management, highlighting the need for
broad differential diagnoses and multidisciplinary
input.
From a therapeutic
perspective, corticosteroids remain the
cornerstone for acute management. However, rates
of relapse and steroid dependence are
considerable, prompting the adoption of B-cell
depletion therapy such as rituximab. This targets
CD20+ B cells and has shown sustained remission
rates in multi-organ IgG4-RD, particularly in
cases with extensive or relapsing disease.
Newer agents
inhibiting T-cell or cytokine signaling including
IL-4/IL-13 and Bruton’s tyrosine kinase inhibitors
are now in clinical trials, reflective of the
shifting understanding of IgG4-RD
pathogenesis.[7,8]
Follow-up is
essential, given the tendency for chronic fibrosis
to progress even after immunosuppressive therapy.
Recent reports now recommend regular clinical
assessment, imaging, and serological monitoring to
optimize long-term outcomes and minimize
irreversible tissue damage.
Conclusion
This case of IgG4-RD
involving a long bone and presenting as an
osteolytic lesion illustrates the inherent
complexity of the disease. It reinforces that
IgG4-RD, though rare in bone, should be considered
in the differential diagnosis of lytic skeletal
lesions with compatible histology and serology.
The literature strongly suggests that early,
accurate diagnosis and a tailored
immunosuppressive approach are critical for good
patient outcomes, especially in atypical
presentations.
Advances in
understanding the cellular and molecular
underpinnings of IgG4-RD particularly the roles of
cytotoxic T cells and targeted biologic therapies
are likely to shape the next generation of
management paradigms. Sustained multidisciplinary
collaboration and continued research into
predictive biomarkers will further enhance care
for patients with this enigmatic and multifaceted
condition.
References
- Nambiar S, Oliver TI. IgG4-Related Disease.
2023 Aug 8. In: StatPearls [Internet]. Treasure
Island (FL): StatPearls Publishing; 2025 Jan.
- Hirabayashi K, Zamboni G. IgG4-related
disease. Pathologica. 2012
Apr;104(2):43-55.
- Nizar, AH, Toubi E. IgG4-related disease: case
report and literature review. Autoimmun
Highlights 2015;6:7–15.
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya
N. High serum IgG4 concentrations in patients
with sclerosing pancreatitis. New England
Journal of Medicine. 2001;344:732–738.
- Umehara H, Okazaki K, Kawa S et al. The 2020
revised comprehensive diagnostic (RCD) criteria
for IgG4-RD. Mod Rheumatol 2021;
31:529–533.
- Deshpande V, Zen Y, Chan JK et al. Consensus
statement on the pathology of IgG4-related
disease. Mod Pathol 2012; 25:1181–1192.
- Perugino CA, Mattoo H, Mahajan VS, Maehara T,
Wallace ZS, Pillai S, Stone JH. Emerging
Treatment Models in Rheumatology: IgG4-Related
Disease: Insights Into Human Immunology and
Targeted Therapies. Arthritis Rheumatol.
2017 Sep;69(9):1722-1732.
- Chen LYC. IgG4-related disease for the
hematologist. Hematology Am Soc Hematol Educ
Program. 2024 Dec 6;2024(1):594-603.
|




