Case Report
Wolfram to Alstrom: Analysis of a Diagnostic Error
Authors:
Annapoorna Chakrabarty, Intern, Kasturba Medical College, Manipal Academy of Higher Education, Manipal, India,
Shipra Rai, Specialist Physician, Prime Medical Centre, Al Qasimia, Sharjah, UAE,
Kavitha Saravu, Head of the Department of Infectious Diseases, Kasturba Hospital, Manipal, India.
Address for Correspondence
Dr. Shipra Rai,
Prime Medical Centre,
Al Qasimia,
Sharjah, UAE
E-mail: dr.shiprarai@gmail.com.
Citation
Chakrabarty A, Rai S, Saravu K. Wolfram to Alstrom: Analysis of a Diagnostic Error. Online J Health Allied Scs.
2021;20(2):8. Available at URL:
https://www.ojhas.org/issue78/2021-2-8.html
Submitted: Jun 1,
2021; Accepted: Aug 5, 2021; Published: Aug 25, 2021 |
|
|
|
Introduction:
Wolfram Syndrome or DIDMOAD is a very rare, autosomal recessive disease with a prevalence of 1 in 100,000-700,000 depending on the clinical features[1]. It is characterized by diabetes insipidus(DI), juvenile onset Type 1 diabetes mellitus (DM), optic atrophy(OA), deafness(D). It is caused by mutations in WSF1 or WSF2 gene. Type 1 DM and optic atrophy are sufficient for diagnosis[2].
Alstrom Syndrome is an autosomal recessive disorder characterized by type 2 diabetes mellitus, cone-rod atrophy and hearing loss. This is caused due to a mutation in the ALMS1 gene on chromosome 2p 13 which codes for a ciliary protein[3]. This leads to multi organ pathologies due to ciliopathy.
Ciliopathy in Alstrom syndrome also makes its symptoms similar to Bardet-Biedl syndrome, which is caused due to a mutation in BBS gene, and presents later in life. The EURO-WABB (Wolfram, Alstrom, Bardet-Biedl) Project was started to document these cases to study their natural history and to prevent misdiagnosis and mismanagement[4].
Case Report
A 19 year old male, known case of Wolfram Syndrome was referred to the hospital with a mass in the left upper quadrant of the abdomen which appeared insidiously and progressively increased in size over 3 months. He also reported two episodes of dark colored urine starting two days before admission to the hospital.
Past history revealed that the patient had had delays in reaching developmental milestones in gross motor, personal-social and language domains. He was evaluated for hearing deficits at age 3 and was diagnosed with sensorineural hearing loss after a brainstem evoked response audiometry (BERA) test. Around the same age he also started to have vision problems for which he was not evaluated. At age 13 he was diagnosed with Type 1 diabetes mellitus (DM) and started on insulin therapy which he continued for the last 5 years. During the same visit, in light of his vision and hearing loss, coupled with Type 1 DM, a diagnosis of Wolfram Syndrome was made. At age 19, the patient presented to a clinic with his current symptoms and was subsequently referred to our hospital.
On examination, he was found to have gross splenomegaly, spleen was felt 16 cm below the costal margin, soft and non-tender. Cardiovascular examination was unremarkable except for the presence of a loud P2 heart sound. The patient was of short stature (149cm) and weighed 49 kg putting his BMI in the normal range (22.07). His head circumference was lower than normal (48.5cm). Fundoscopy revealed diffuse retinal pigment epithelial (RPE) atrophy and attenuation of retinal vasculature.
Blood investigations showed low hemoglobin (5.3g/dL) and hematocrit (18%), decreased RBC indices and increased red cell distribution width, low platelet count (3.2x103/microliter), low total WBC count (2.8x103/microliter). In addition the patient had high fasting blood glucose (362g/dL) and HbA1c (9.1%), hypertriglyceridemia (319mg/dL) and elevated total and direct bilirubin levels. Urine examination showed glycosuria. Ultrasonography revealed chronic hepatic parenchymal changes and features of portal hypertension with dilated splenic vein (13mm) and gross splenomegaly (21cm along the anteroposterior axis). Chest X-Ray (Fig.1) showed prominent main pulmonary artery, loss of pulmonary conus and biatrial enlargement as evidenced by the double contour within the right atrial shadow. Cardiac apex is round and elevated above the diaphragm suggesting right ventricular hypertrophy.
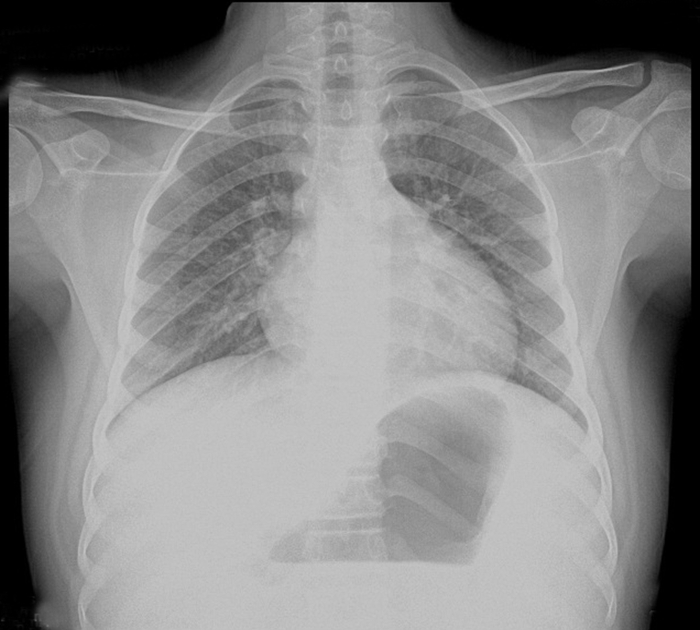 |
Fig.1: Chest X-Ray showing evidence of right ventricular hypertrophy |
The clinical evidence of ineffective glycemic control with insulin, dyslipidemia and multiorgan involvement led to revision of the diagnosis to Alstrom syndrome.
Discussion
Overlap in the clinical presentation of these two syndromes reveals important challenges in the diagnosis of rare diseases. DM is a feature of both conditions but early age of presentation led to the patient being misdiagnosed with Type 1 DM. This along with loss of vision and sensorineural hearing loss led to the diagnosis of Wolfram Syndrome without confirmatory genetic testing due to lack of resources. But when patient presented at age 19, with uncontrolled diabetes despite five years of insulin therapy, the diagnostic error was identified and corrected. The treatment plan was revised for Type 2 DM by adding oral hypoglycemics as well as rapid acting insulin after which blood glucose levels were controlled [5]. Clinical evidence for insulin resistance such as acanthosis nigricans is usually the key to detecting Type 2 DM in juvenile presentation[6]. Therefore, in young children presenting with syndromic features and symptoms of diabetes, it is important to perform a thorough clinical examination to avoid mismanagement.
Sensorineural hearing loss is also seen in both conditions, and apart from subtle differences in age of presentation, does not aid in differentiation. Similarly, growth retardation and developmental delay are features of both Alstrom and Wolfram syndrome and cannot be used in this case to support one diagnosis over the other. Vision loss in this patient due to retinal pigment epithelium atrophy more closely resembles the cone-rod atrophy found in Alstrom syndrome than the optic atrophy characteristic of Wolfram.
Childhood truncal obesity is a characteristic feature of Alstrom syndrome which was absent in this patient. This could be explained by the presence of uncontrolled Type 2 DM leading to weight loss. Juvenile hypertriglyceridemia is a characteristic feature of Alstrom syndrome. The patient was advised a low fat diet and prescribed statins in order to reduce triglyceride levels and prevent further complications like pancreatitis.
Liver parenchymal coarsening on USG is caused due to ciliopathic effect of mutation in the ALSM1 gene which in turn leads to features of portal hypertension. Splenomegaly results from portal hypertension leading to hypersplenism. This explains the low platelet and red cell counts. Iron supplementation was initiated as he had low serum iron, ferritin and TIBC. Splenectomy was advised after surgical consultation, but the patient was not willing to undergo surgery. Loud P2 heart sound and chest X-ray findings are due to pulmonary hypertension caused by lung fibrosis ultimately leading to right ventricular hypertrophy. This is a serious complication which could not have been predicted with the diagnosis of Wolfram syndrome and has therefore increased morbidity in this patient. Dilated cardiomyopathy is another feared complication of Alstrom syndrome and it is important to be on the lookout for impaired cardiac function in long term care.
In further management, renal involvement was ruled out; abdominal ultrasound showed normal kidneys and renal function tests were within normal limits. Genetic testing was advised to confirm diagnosis which the patient declined due to financial constraints.
Conclusion
The only way to definitively diagnose Wolfram or Alstrom syndrome is through gene testing. There are few medical centers in India that offer such facilities and many patients are unable to afford these tests. This case is an eye opener regarding the dearth of affordable diagnostic resources for rare genetic diseases, therefore it is important that we focus on diagnosing such conditions by correctly interpreting the clinical features. The diagnosis of Type 2 diabetes in the paediatric age-group was a particularly tricky aspect of the case. Even though the management of such diseases is primarily dependent on monitoring and treating the symptoms as and when they present [7], gene testing and a definitive diagnosis will help us predict the inheritance of the disease in future generations leading to early diagnosis and treatment and a better quality of life for the patient.
Conflict of Interest: None
References
- Viswanathan V, Medempudi S, Kadiri M. Wolfram syndrome. JAPI. 2008 Mar 14;56:197-9.
- Ari S, Keklíkçí U, Çaça I, Ünlü K, Kayabasi H. Wolfram syndrome: case report and review of the literature. Comprehensive Therapy. 2007 Sep 1;33(1):18-20.
- Marshall DJ, Maffei P, Collin BG,Naggert KJ. Alstrom syndrome: genetics and clinical overview. Current Genomics. 2011 May 1;12(3):225-35.
- Farmer A, Aymé S, de Heredia ML, et al. EURO-WABB: an EU rare diseases registry for Wolfram syndrome, Alström syndrome and Bardet-Biedl syndrome. BMC Pediatrics. 2013 Dec;13(1):130.
- DeFronzo RA. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009 Apr 1;58(4):773-95.
- Bakar AA, Kamal NM, Alsaedi A, Turkistani R, Aldosari D. Alström syndrome: A novel mutation in Saudi girl with insulin-resistant diabetes. Medicine. 2017 Mar;96(10).
- Marshall JD, Beck S, Maffei P, Naggert JK. Alström syndrome. European Journal of Human Genetics. 2007 Dec;15(12):1193.
|

