Case Report
Extra Nodal (Cutaneous) Rosai Dorfman Disease
Authors:
Talha Ahmed, Junior Resident,
Jayarama Shenoy, Professor,
Ashfaque Mohammed, Professor,
Syed Mahmood Ayaz, Assistant Professor,
Bichu Joseph Maliakal, Assistant Professor,
Manmohan Singh Rajput, Senior Resident,
Department of Surgery, Kasturba Medical College, Mangalore, Manipal Academy of Higher Education, Karnataka, India.
Address for Correspondence
Dr. Ashfaque Mohammed,
Professor,
Department of Surgery,
Kasturba Medical College, Mangalore,
Manipal Academy of Higher Education,
Karnataka, India.
E-mail: ashfaque.mohammed69@gmail.com.
Citation
Ahmed T, Shenoy J, Mohammed A, Ayaz SM, Malaikal BJ, Rajput MS. Extra
Nodal (Cutaneous) Rosai Dorfman Disease. Online J Health Allied Scs.
2020;19(2):15. Available at URL:
https://www.ojhas.org/issue74/2020-2-15.html
Submitted: Apr 25,
2020; Accepted: Sep 1, 2020; Published: Sep 20, 2020 |
|
|
|
Background
Rosai Dorfman disease, also known as Sinus histiocytosis with massive lymphadenopathy (SHML) was first described by Destombes in 1965 and then subsequently categorized as a distinct clinicopathological disorder in 1969 by Juan Rosai and Ronald Dorfman (1). It is a benign, self-limiting disease of unknown etiology occurring mostly in young patients, characterized by proliferation and accumulation histiocytes in lymph nodes or extra nodal sites such as skin, nasal sinuses, central nervous system, kidney and gastrointestinal tract. The usual presentation is a lymph nodal swelling in the cervical region associated with constitutional symptoms such as fever and elevated ESR with leukocytosis. Altered immune responses and infectious agents are thought to play a role in its occurrence. The hallmark of the disease is emperipolesis, which is the phagocytosis of lymphocytes or erythrocytes by histiocytes and this is required for the diagnosis (1).
Case Presentation
This is the case of a 28-year-old South Indian female who presented to Government Wenlock Hospital, Mangalore with a painless lump on the left anterior aspect of the arm for 2 years, which was progressively increasing in size. She had no preceding history of trauma, fever or any similar swellings elsewhere. She denied any constitutional symptoms or neurological deficits. She had no associated history of weight loss. On examination, it was a firm, well-defined swelling on the ventral aspect of the left arm, 6x4cm in size which was mobile in all planes, present 4cm below the acromion process and 10cm above cubital fossa of the left arm, with no associated skin changes, discharge or erythema over the swelling. The skin could not be pinched over the swelling and the swelling was not attached to any deeper structures, which was suggestive of a cutaneous swelling. There was no associated lymphadenopathy. Other systems examined were normal clinically and on imaging.
Preoperatively, a full blood count was done which revealed values within normal parameters. An Ultrasound scan of the breast and axilla were done to rule out primary malignancy, which was negative for any lesions.
Local Ultrasound of the arm was done which showed large heterogeneous lesion over the anteromedial aspect of the left arm measuring 6.5x3.5cm in the superficial plane. Another 3.2x2.1cm heterogeneous lesion in the posterolateral aspect of the left upper arm in the superficial plane. Features were likely suggestive of soft tissue tumors in the left arm.
Fine Needle Aspiration Cytology was done to further aid the diagnosis which showed a moderately cellular smear with fragments of spindle cells with scant to moderate cytoplasm, oval to waxy vesicular nuclei. Background shows dense polymorphous population of lymphocytes comprising of small mature and activated forms and few histiocytes with ingested necrotic debris and occasional plasma cells in a fatty background. The provisional diagnosis given were:
A) Inflammatory lesion likely to be fungal/parasitic.
B) Inflammatory cell rich benign/intermediate grade with soft tissue neoplasm – Benign neural neoplasm/Inflammatory myofibroblastic tumor.
The patient underwent wide local excision including excision of the involved skin which was done under general anesthesia and intra-op findings revealed a firm swelling in the subcutaneous plane with adhesions to the overlying fat but not involving the underlying muscle.
On cut section, it was a well circumscribed solid grey white homogenous lesion with specks of hemorrhage and few yellow areas abutting all resected margins. Histopathology revealed dense cellular infiltrate involving dermis and subcutaneous tissue made up of aggregates of lymphocytes and sheets of large, pale histiocytes with large round vesicular nuclei arranged in a nodular, lobular pattern, with emperipolesis in some. At places the lymphocytic nodules contained numerous plasma cells along with few neutrophilic micro abscesses and stromal fibrosis seen in some places. These features are suggestive of cutaneous (extra nodal) Rosai Dorfmann disease. Immunohistochemistry studies revealed the specimen to be positive for CD68 and S100.
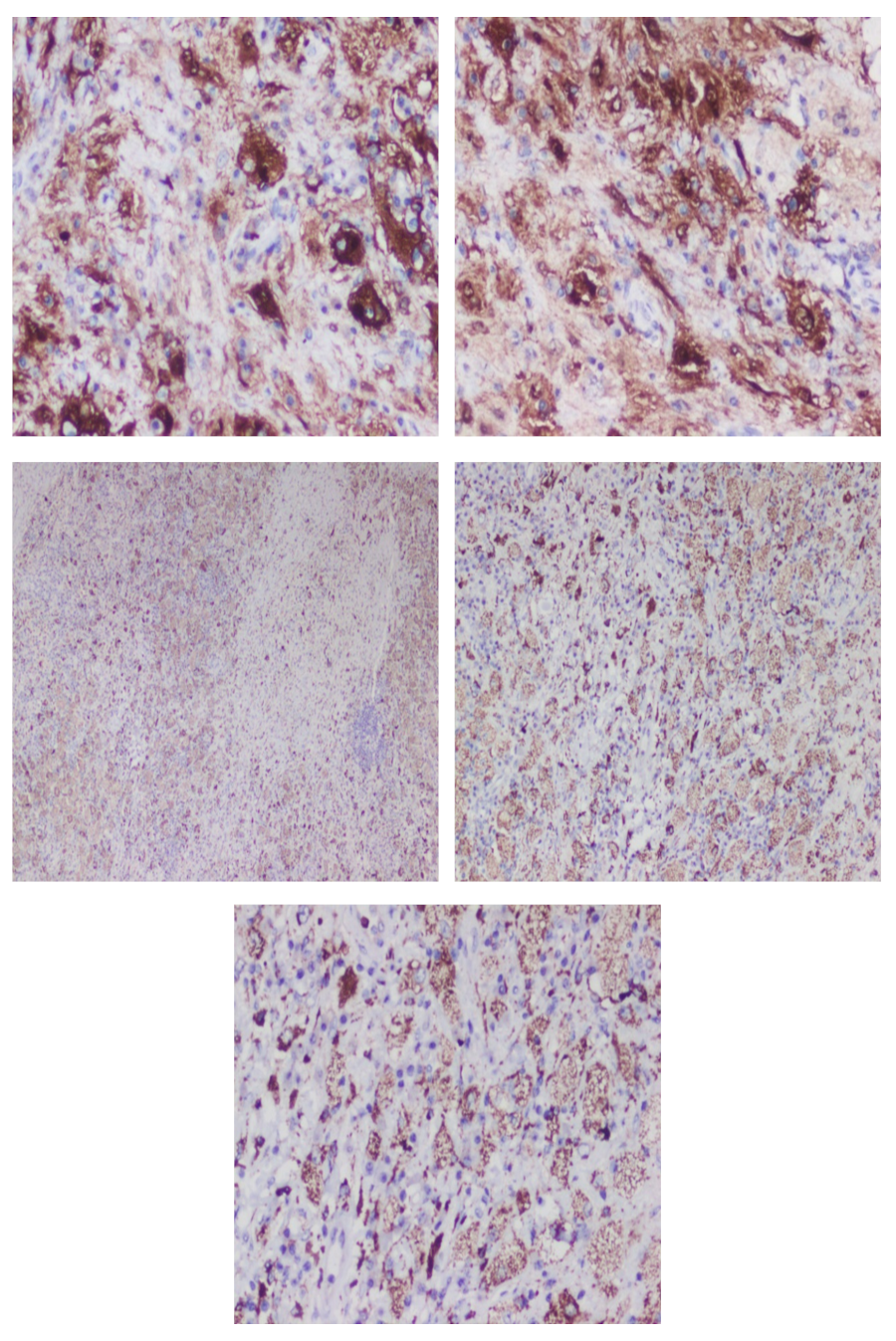 |
Figure 1: Histopathology examination of left anterior arm mass demonstrating characteristic emperipolesis and immunohistochemistry CD68 and S100 positive |
Postoperatively there was no discernible nerve damage with resulting loss of power or cutaneous sensation.
In 2 months of follow up, there was no evidence of recurrence at the site of lesion and no swellings elsewhere on the body.
Discussion
Rosai Dorfman disease is a class II histiocytosis, a rare inflammatory non-neoplastic disease with a worldwide distribution and 80% cases occurring in children and young adults in the 2nd and 3rd decade of life with a slight male predominance of 58% (2). It is not usually life threatening and most often does not require therapy apart from surgical excision which is considered curative; but it may undergo exacerbations and remissions. It is of unknown etiology, but studies reveal that Human Herpes Virus 6, Parvovirus B19 and Ebstein Barr Virus to have a role in the pathogenesis (1). However, the etiology is still presumed to be multifactorial and many patients have allergic and autoimmune diseases such as asthma, hemolytic anemias, rheumatoid arthritis and systemic lupus erythematosus (2).
Rosai Dorfman disease can be either nodal or extra nodal, with the latter occurring in 43% of cases (3). In the largest study of Rosai Dorfman disease ever conducted, 182 (43%) of the 423 patients evaluated had extra nodal disease, and only 13 (3%) presented with the typical morphological profile of soft tissue lesion without detectable lymphadenopathy (4). The most commonly affected lymph node groups are cervical, followed by inguinal, axillary and mediastinal. It is very seldom fatal, and fatality results from extension of the disease into vital organs with impaired function of the same. The extra nodal form of the disease can involve skin most commonly, as well as paranasal sinuses, respiratory and gastrointestinal tract and retro-orbital and central nervous system tissues. The cutaneous form comprises of 1/3rd of extra nodal cases and it involves mostly the head and neck region.
A cutaneous-only form of Rosai Dorfman disease (CRDD) has been identified with some researchers suggesting that it may be a different clinicopathological entity from the nodal form. CRDD seems to have preponderance for the female population and particularly for those of Asian descent. Our patient falls under the CRDD category. In CRDD, patients typically present with normal laboratory data and no lymphadenopathy with varying lesions ranging from 1-30cm in size (1). Clinical features of CRDD are variable and can include asymptomatic yellow-red to brownish nodular papules or plaques with facial skin being most commonly affected (5). There is no real use for further workup such CT imaging for spread in CRDD (1).
Due to the self-limiting nature and the rarity of the disease, a definite treatment protocol has not been established thus far. However, for patients with CRDD such as ours, excision of the mass has proved to be curative in most cases with negligible rates of recurrence. The proposed treatment algorithm for other forms of the disease is shown in Fig 2.
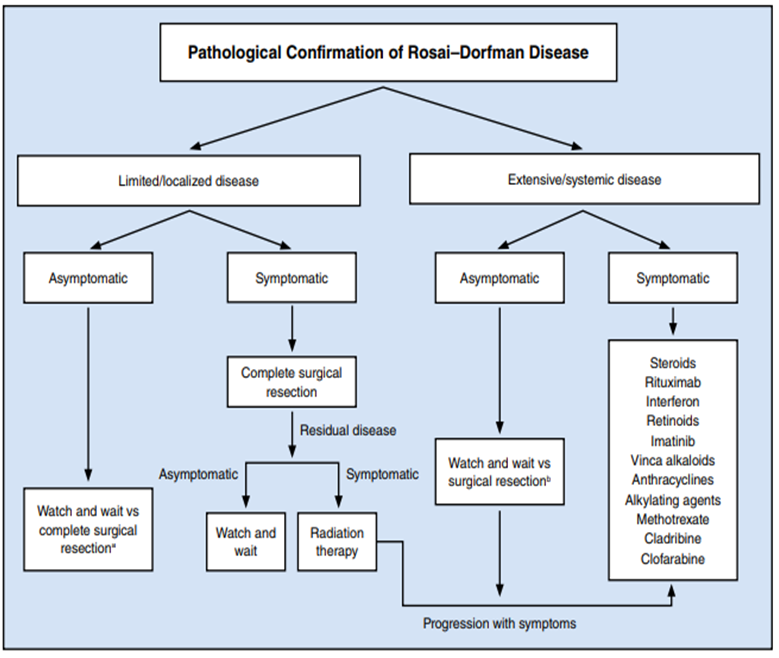 |
Figure 2: Treatment algorithm for Rosai–Dorfman disease. (1) |
Conclusions
Rosai Dorfman disease is a rare non-malignant histiocytic disorder which can be of nodal or extra nodal type. In our patient it was of the extra nodal cutaneous type, which was managed with surgical excision. The patient is on monthly follow up subsequently to watch out for recurrence at the site of excision or elsewhere.
References
- Dalia S, Sagatys E, Sokol L, Kubal T. Rosai–Dorfman Disease: Tumor Biology, Clinical Features, Pathology, and Treatment. Can Con. Oct 2014;21(4). Available at https://journals.sagepub.com/doi/pdf/10.1177/107327481402100408
- Komaragiri M, Sparber LS, Santos-Zabala ML, Dardik
M, Chamberlain RS. Extranodal Rosai–Dorfman disease: a rare soft tissue neoplasm masquerading as a sarcoma.
Wor Jou of Surg Onc. 2013;11:63. Available at http://www.wjso.com/content/11/1/63
- Costa ALP, Silva NO, Motta MP, Athanazio RA, Athanazio DA, Athanazio PRF. Soft tissue Rosai-Dorfman disease of the posterior mediastinum.
J Bras Pneumol. 2009;35(7):717-720. Available at http://www.scielo.br/pdf/jbpneu/v35n7/v35n7a15.pdf
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity.
Semin Diagn Pathol. 1990;7(1):19-73
- Hinojosa T, Ramos E, Lewis DJ, del Angel L, Vangipuram
R, Peranteau AJ, Tyring SK. Cutaneous Rosai-Dorfman disease: A separate clinical entity.
Jour of Derm Derm Surg. July 2017;21(2):107-109.
|

